RNA-seq在subread比对时提示不能找到gene identifie 同时输出的txt文件格式混乱
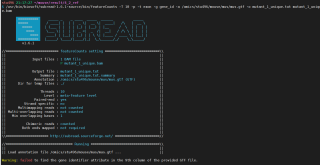
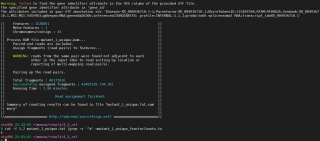
我在利用subread软件进行counts数计算的时候报了warning,但程序还能继续运行
导出txt文件后发现里面很乱,所有的结果都混在一行
想请教是哪一步出问题了 非常感谢

```shell
下面是使用的代码
/usr/bin/biosoft/subread-1.6.1-source/bin/featureCounts -T 10 -p -t exon -g gene_id -a /omics/stu496/mouse/mus/mus.gtf -o mutant_1_unique.txt mutant_1_unique.bam
/omics/stu496/mouse/mus/mus.gtf是之前在ncbi上下载的gff文件
mutant_1_unique.bam是用tophat对fastq文件比对后的bam文件
``
1、警告信息 "Unable to find gene identifier for a feature" 表示 Subread 在处理 GTF 文件时无法找到 gene_id 属性,也就是说你使用的 GTF 文件中缺少了 gene_id 列。这可能是 GTF 文件格式有问题,或者你使用的 GTF 文件版本与 Subread 软件不兼容。建议检查 GTF 文件是否有语法错误,或者尝试使用不同的 GTF 文件或 Subread 版本重新进行分析。
2、如果导出的 txt 文件内容混乱,可能是因为 featureCounts 工具在输出结果时出现了问题。建议检查 featureCounts 工具的输出格式是否有误,或者尝试使用其他工具(例如 HTSeq)来进行基因组计数。
3、还有一些其他可能的原因,也可能导致类似的问题:
①GTF 文件的路径不正确,featureCounts 工具无法找到 GTF 文件
②GTF 文件格式不正确,导致 featureCounts 工具无法正常解析
③bam 文件格式不正确,导致 featureCounts 工具无法正常读取
④其他软件问题,例如 Subread 版本问题、操作系统问题等
你这几张图,无法查看,你应该是放入了代码块中,应该是直接粘贴出来就好
你可参考下这个实例【RNA_seq(1)植物转录组实战(中)之subread工具进行序列比对和转录组生物学定量】,链接:https://blog.csdn.net/Candle_light/article/details/81953107
看下是否有哪些地方不同
RNA-seq中遇到的问题(1)
借鉴下
https://blog.csdn.net/twistti/article/details/120091524