在为'head'函数选择方法时评估'x'参数出了错: 'x' must be atomic ?
RStudio:
> filtered_seurat
An object of class Seurat
37256 features across 14622 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
> split_seurat <- SplitObject(filtered_seurat, split.by = "orig.ident")[1:8]
> split_seurat
$lbm1
An object of class Seurat
37256 features across 4610 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lbm2
An object of class Seurat
37256 features across 1696 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lbm3
An object of class Seurat
37256 features across 5890 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lbm4
An object of class Seurat
37256 features across 982 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lc1
An object of class Seurat
37256 features across 381 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lc2
An object of class Seurat
37256 features across 351 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lc3
An object of class Seurat
37256 features across 331 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
$lc4
An object of class Seurat
37256 features across 381 samples within 1 assay
Active assay: RNA (37256 features, 0 variable features)
> split_seurat <- lapply(X = split_seurat, FUN = SCTransform)
> split_seurat
$lbm1
An object of class Seurat
48939 features across 4610 samples within 2 assays
Active assay: SCT (11683 features, 3000 variable features)
1 other assay present: RNA
$lbm2
An object of class Seurat
49760 features across 1696 samples within 2 assays
Active assay: SCT (12504 features, 3000 variable features)
1 other assay present: RNA
$lbm3
An object of class Seurat
47758 features across 5890 samples within 2 assays
Active assay: SCT (10502 features, 3000 variable features)
1 other assay present: RNA
$lbm4
An object of class Seurat
48461 features across 982 samples within 2 assays
Active assay: SCT (11205 features, 3000 variable features)
1 other assay present: RNA
$lc1
An object of class Seurat
55730 features across 381 samples within 2 assays
Active assay: SCT (18474 features, 3000 variable features)
1 other assay present: RNA
$lc2
An object of class Seurat
55389 features across 351 samples within 2 assays
Active assay: SCT (18133 features, 3000 variable features)
1 other assay present: RNA
$lc3
An object of class Seurat
56942 features across 331 samples within 2 assays
Active assay: SCT (19686 features, 3000 variable features)
1 other assay present: RNA
$lc4
An object of class Seurat
56121 features across 381 samples within 2 assays
Active assay: SCT (18865 features, 3000 variable features)
1 other assay present: RNA
> features <- SelectIntegrationFeatures(split_seurat)
Error in h(simpleError(msg, call)) :
在为'head'函数选择方法时评估'x'参数出了错: 'x' must be atomic
如上,单细胞测序数据,出错之后,运行:
> is.atomic(split_seurat)
[1] FALSE
> is.list(split_seurat)
[1] TRUE但是使用unlist(split_seurat)后,仍然出现:
> is.atomic(split_seurat)
[1] FALSE
> is.list(split_seurat)
[1] TRUE
运行SelectIntegrationFeatures() 还是报错。
接下来使用内置数据进行测试:
> devtools::install_github('satijalab/seurat-data')
> library(SeuratData)
> AvailableData()
> InstallData("panc8")
> data("panc8")
> panc8
An object of class Seurat
34363 features across 14890 samples within 1 assay
Active assay: RNA (34363 features, 0 variable features)
> pancreas.list <- SplitObject(panc8, split.by = "tech")[1:2]
> pancreas.list
$celseq
An object of class Seurat
34363 features across 1004 samples within 1 assay
Active assay: RNA (34363 features, 0 variable features)
$celseq2
An object of class Seurat
34363 features across 2285 samples within 1 assay
Active assay: RNA (34363 features, 0 variable features)
> pancreas.list <- lapply(X = pancreas.list, FUN = SCTransform)
> pancreas.list
$celseq
An object of class Seurat
51110 features across 1004 samples within 2 assays
Active assay: SCT (16747 features, 3000 variable features)
1 other assay present: RNA
$celseq2
An object of class Seurat
51501 features across 2285 samples within 2 assays
Active assay: SCT (17138 features, 3000 variable features)
1 other assay present: RNA
> features <- SelectIntegrationFeatures(pancreas.list)
以上测试数据集并没有报错,但是运行:
> is.atomic(pancreas.list)
[1] FALSE
> is.list(pancreas.list)
[1] TRUE
is.atomic(pancreas.list) 竟然也是FALSE。
不解。。。
看不懂
*望采纳**谢谢 参考帮助文档:对于cor.test来说,输入变量x,y必须是:numeric vectors of data values,所以当你输入两个表的时候就会报错; with(mtcars,cor.test(cyl,disp))##这个可以运行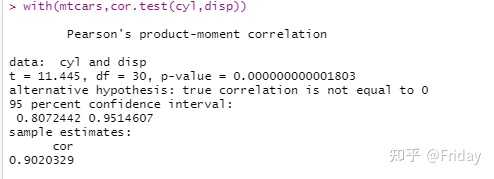 cor.test(mtcars,mtcars)##这个则报错
cor.test(mtcars,mtcars)##这个则报错 如果是要查看一个数据框内不同变量的之间的相关性,在每个变量都是数值型的情况下是可行的cor(mtcars,mtcars)
如果是要查看一个数据框内不同变量的之间的相关性,在每个变量都是数值型的情况下是可行的cor(mtcars,mtcars)