lammps做剪切过程中温度变化很大怎么办(语言-c++)
最近在用LAMMPS进行DEM模拟。大约2000个粒子被放置在一个固定体积V的周期盒子中。通过Lees-Edwards边界条件,粒子在剪切速率下进行均匀稳定的简单剪切。势函数考虑hooke(参考命令:pair_style granular或者pair_style hooke)
剪切过程中,温度远远高于设定的温度值,请问各位这是什么原因导致的呢,是不是我的in文件有问题?
试过直接设定想要的盒子尺寸(2000个粒子,体积分数为0.51,直径为1,盒子的尺寸为12.712.712.7),而不加change_box all x scale 0.5 y scale 0.5 z scale 0.5,并且只计算比较小的剪切速率,相当小时温度可以稍微降下来,但是插入的粒子数达不到2000。并且想算较大的剪切速率的还是不行。
以下是我的in文件
以下是我的in文件
################################################
variable d equal 1.0
variable rc equal 1.25*${d}
variable density equal 1.0
variable H equal 25.42
variable L equal 25.42
############ 模型初始化(单位:微米)#############
units micro
boundary p p p
atom_style sphere
region box block 0 ${L} 0 ${L} 0 ${H}
create_box 1 box
create_atoms 1 random 2000 12345 NULL overlap 1 maxtry 500
neighbor 0.2 bin
neigh_modify every 1 delay 0 check yes
############ 设置颗粒属性 ##########
set atom * diameter ${d} density ${density}
############ 势函数参数 #############
variable kn equal 1e4
variable kt equal 3e3
variable e equal 0.7
variable miu equal 0.1
variable shearvel equal 0.02
variable dt equal 2e-5
variable steps1 equal 1000
variable steps2 equal 10000
variable steps3 equal 1000
variable steps4 equal 50000
############ 势函数 ############
pair_style granular
pair_coeff * * hooke ${kn} ${e} tangential linear_history ${kt} 0.0 ${miu} damping tsuji
comm_modify vel yes
############ 温度初始化 ############
velocity all create 300 1231
########################################
compute s all stress/atom NULL
compute p all reduce sum c_s[1] c_s[2] c_s[3]
variable stressx equal c_p[1]/(vol)
variable stressy equal c_p[2]/(vol)
variable stressz equal c_p[3]/(vol)
variable press equal -(c_p[1]+c_p[2]+c_p[3])/(3*vol)
############# 驰豫 #####################
fix 1 all npt temp 300.0 300.0 $(10*dt) iso 0 0 1000 drag 2
fix 2 all momentum 1 linear 1 1 1
dump 1 all custom 5000 npt1-hooke.xyz id type x y z
thermo_style custom step temp press
thermo 100
timestep ${dt}
reset_timestep 0
run ${steps1}
unfix 1
unfix 2
undump 1
############## 剪切 #####################
change_box all x scale 0.5 y scale 0.5 z scale 0.5
change_box all triclinic
fix 1 all nvt/sllod temp 300 300 $(100*dt)
fix 2 all deform 1 xz erate ${shearvel} units box remap v
fix stress all print 100 "${press} ${stressx} ${stressy} ${stressz}" file stress-hooke.dat screen no
dump 2 all atom 100 dump-hooke.shear
dump 1 all custom 10 shear-hooke.xyz id type x y z vx vy vz fx fy fz diameter
timestep ${dt}
reset_timestep 0
run ${steps4}
#min_style cg
minimize 1e-4 1e-6 10000 10000
这个问题很可能是因为模拟条件不适当导致的。有几点可以考虑改进:
考虑使用不同的随机初始速度来初始化粒子,例如:velocity all create 300 1231,可以尝试使用其他速度,例如:velocity all create 300 123
可以适当调整 NPT 温度调节的参数,例如增加 drag 值,或者增加控制温度的频率。
如果剪切速率较大,可以考虑使用 Verlet 数值积分,而不是 velocity-Verlet 数值积分。
可以尝试使用不同的粒子间作用力(如 Hertz-Mindlin 或者 Mohr-Coulomb 粒子间作用力)来替换默认的 Hooke 粒子间作用力。
这些都是可以考虑的改进方法,但并不能保证解决问题,因为还需要根据具体情况进行调整。
方法一
摩擦力和滚动摩擦力(参考命令:fix 1 all gran/hooke history $mu $mu_r)。
LAMMPS can be used to perform DEM simulations. Approximately 2000 particles are placed in a fixed volume V periodic box. Through Lees-Edwards boundary conditions, particles are uniformly and stably sheared at shear rate. The potential function considers hooke (reference command: pair_style granular or pair_style hooke), friction force and rolling friction force (reference command: fix 1 all gran/hooke history $mu $mu_r).
方法二
这可能是由于周期性边界条件引起的。由于粒子受到周期性的力,因此激活的能量会远大于静态的力的能量,从而导致温度上升。解决这一问题的一个方法是采用非周期性的边界条件,或者改变剪切速率,以降低激活的能量。此外,也可以使用质量较小的粒子,以减少激活的能量,或者采用随机踊跃来模拟热激活。
方法三
这可能是由于模拟参数未正确设定或模拟流体(如气体)过度加热导致的。正确设定模拟参数可以避免过度加热。例如,可以使用刚体时间步长和相互作用力参数来减少加速度变化量。另外,使用动能削减方法来缓解加热也是有效的。同时,应避免在模拟中使用太强的耦合,例如使用过大的Nose-Hoover系数(thermo_nh),可能会导致过度加热。
题主,体系的温度并不能完全按照以上公式进行计算吧?
一般来说,流体的温度计算公式中应该去掉流体的流动速度。
在lammps中,有两个命令可实现这个功能:
第一种是 : compute temp/partial
该命令语法为:
compute ID group-ID temp/partial xflag yflag zflag
其中,group-ID为需要计算温度的原子组。
xflag yflag zflag确定是否计算该方向的温度。取值为1表示计算温度是包含该方向的速度,取值为0表示不包含该方向的速度。
如流体沿y方向流动,在计算流体温度时,不包含y方向速度,只计算x和z方向速度:
compute myTemp flow temp/partial 1 0 1
第二种: compute temp/com
该命令首先计算原子组质心的速度,在计算温度时,去除质心速度。
使用方法如下:
compute myTemp mobile temp/com
重新计算温度后,可在thermo或者fix命令中使用新的命令,如用在nvt系综设置中:
fix 1 flow nvt temp 300 300 0.1
fix_modify 1 temp myTemp
或者:
thermo_modify temp myTemp
对比下这个相对完整的实例:lammps剪切模拟关键技术讲解,链接:https://blog.csdn.net/lammps_jiayou/article/details/121605813
不一定,高温可能是由多种原因引起的。
高频碰撞:LAMMPS中默认的碰撞模型可能导致粒子之间发生高频碰撞,从而提高温度。
内力过大:如果势函数设置不当,特别是对于Hooke势函数,它们可能产生过大的内力,从而提高温度。
缺乏热力学控制:如果您的模拟没有考虑热力学控制,则温度可能升高。
时间步长过大:如果时间步长过大,则可能导致粒子运动不稳定,从而提高温度。
建议您细心检查您的输入文件,特别是势函数、热力学控制和时间步长,确保这些因素是合适的。此外,您可以尝试不同的碰撞模型或更改时间步长来验证结果。
高于预期的温度可能是由多种因素导致的,其中一些可能是:
无效的温度控制方法:确保使用适当的温度控制方法(例如NPT或NVT),以确保温度处于预期范围。
不合适的初始温度:将温度设置为不适当的值,也可能导致结果不正确。
模拟参数:使用不适当的模拟参数,例如时间步长或摩擦系数,也可能导致错误结果。
势函数:不合适的势函数也可能导致不正确的结果。请确保选择适当的势函数,并确保与模拟条件相符。
因此,请检查您的 in 文件,确保使用了适当的模拟参数,以及检查温度控制方法和势函数是否正确。
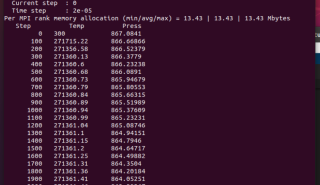
你把运行日志发给我看一下
剪切过程中温度过高的原因可能有很多,可能与模拟参数设置、物理模型选择等有关,具体需要根据您的实验条件和实验结果来分析。
以下是一些可能的原因:
1.模拟时间过短。温度是一个瞬时值,可能会有一定的波动。如果模拟时间不够长,温度的波动会比较大,导致温度偏高。
2.粒子碰撞过于频繁。如果剪切速率过大,粒子之间的碰撞就会变得更加频繁,这会导致温度升高。
3.势函数的选择。不同的势函数具有不同的物理意义和表现形式,可能会对温度的变化产生影响。
4.模拟参数的设置。例如,时间步长、温度控制方式、计算温度的方法等都可能会对温度的变化产生影响。
根据您提供的in文件,无法确定温度高于设定温度的具体原因。您可以检查以下问题:
1.初始化温度是否正确,是否为300K?
2.可以检查初始动能是否正确,以确保初始温度正确设置。
3.剪切速率是否合理,剪切速率过高可能导致温度过高。
4.势函数参数是否合理,例如kn和kt的值是否正确。
如果您已经尝试过减小剪切速率和盒子的尺寸并且温度有所下降,那么可能是由于模拟参数的选择或其他原因导致温度高于设定温度。您可以尝试调整参数或尝试不同的方法以解决这个问题。
目前通过查文献,认为很大可能是边界条件有问题,传统的周期性边界条件无法精确计算剪切流,尤其是剪切速度增大以后,文献中提出一种新的周期性边界,在速度梯度方向施加,一个粒子跑出边界之后,新补上的粒子的速度和位置需要重新计算,称为lees-edward边界条件,lammps中是否能直接设置这种不常规的边界条件,还是说需要改源代码?若能解决,一定酬谢,拜托拜托!